Selected Publications:

|
Ancestral sequence reconstruction of Mic60 reveals a residue signature supporting respiration in yeast
Friederike M. C. Benning*, Tristan A. Bell*, Tran H. Nguyen*, Della Syau, Louise B. Connell, Corrie J. B. daCosta, Luke H. Chao bioRxiv 2024.04.26.591372 2024.04.26.591372; doi: 10.1101/2024.04.26.591372 *equal contribution In eukaryotes, the essential process of cellular respiration takes place in the cristae of mitochondria. The protein Mic60 is known to stabilize crista junctions; however, how the C-terminal Mitofilin domain of Mic60 mediates cristae-supported respiration remains elusive. Here, we used ancestral sequence reconstruction to generate Mitofilin ancestors up to and including the last opisthokont common ancestor (LOCA). We found that yeast-lineage derived Mitofilin ancestors as far back as the LOCA rescue respiration. By comparing Mitofilin ancestors with different respiratory phenotypes, we identify four residues that explain the difference between respiration functional yeast- and non-functional animal-derived common Mitofilin ancestors. Our results imply that Mitofilin-supported respiration in yeast stems from a conserved mechanism, and provide a foundation for investigating the divergence of candidate crista junction interactions present during the emergence of eukaryotes. |
|
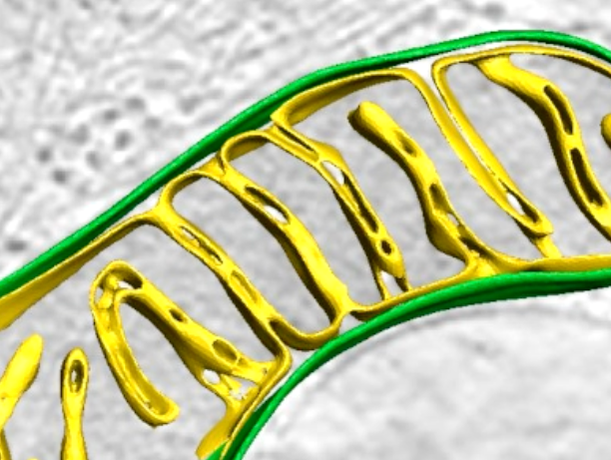
In situ architecture of Opa1-dependent mitochondrial cristae remodeling
Michelle Y. Fry*, Paula P. Navarro*, Xingping Qin, Zintes Inde, Virly Y. Ananda, Camila Makhlouta Lugo, Pusparanee Hakim, Bridget E. Luce, Yifan Ge, Julie L. McDonald, Ilzat Ali, Leillani L. Ha, Benjamin P. Kleinstiver, David C. Chan, Kristopher Sarosiek, Luke H. Chao EMBO Journal 2024 Jan 15; doi: 10.1038/s44318-024-00027-2 *equal contribution PMID: 38225406 Cristae membrane state plays a central role in regulating mitochondrial function and cellular metabolism. The protein Optic atrophy 1 (Opa1) is an important crista remodeler that exists as two forms in the mitochondrion, a membrane-anchored long form (l-Opa1) and a processed short form (s-Opa1). The mechanisms for how Opa1 influences cristae shape have remained unclear due to the lack of native 3D views of cristae morphology. We perform in situ cryo-electron tomography of cryo-focused ion beam milled mouse embryonic fibroblasts to understand how each form of Opa1 influences cristae architecture. In our tomograms, we observe elongated mitochondria with a stacking phenotype, and an absence of tubular cristae, when only l-Opa1 is present. In contrast, when mitochondria contain mainly s-Opa1, we observe irregular cristae packing, an increase in globular cristae, and decreased matrix condensation. Notably, we find the absence of l-Opa1 results in mitochondria with wider cristae junctions. BH3 profiling reveals that absence of l-Opa1 reduces cytochrome c release in response to pro-apoptotic stimuli. We discuss the implications Opa1-dependent cristae morphologies in cell death initiation. |
|
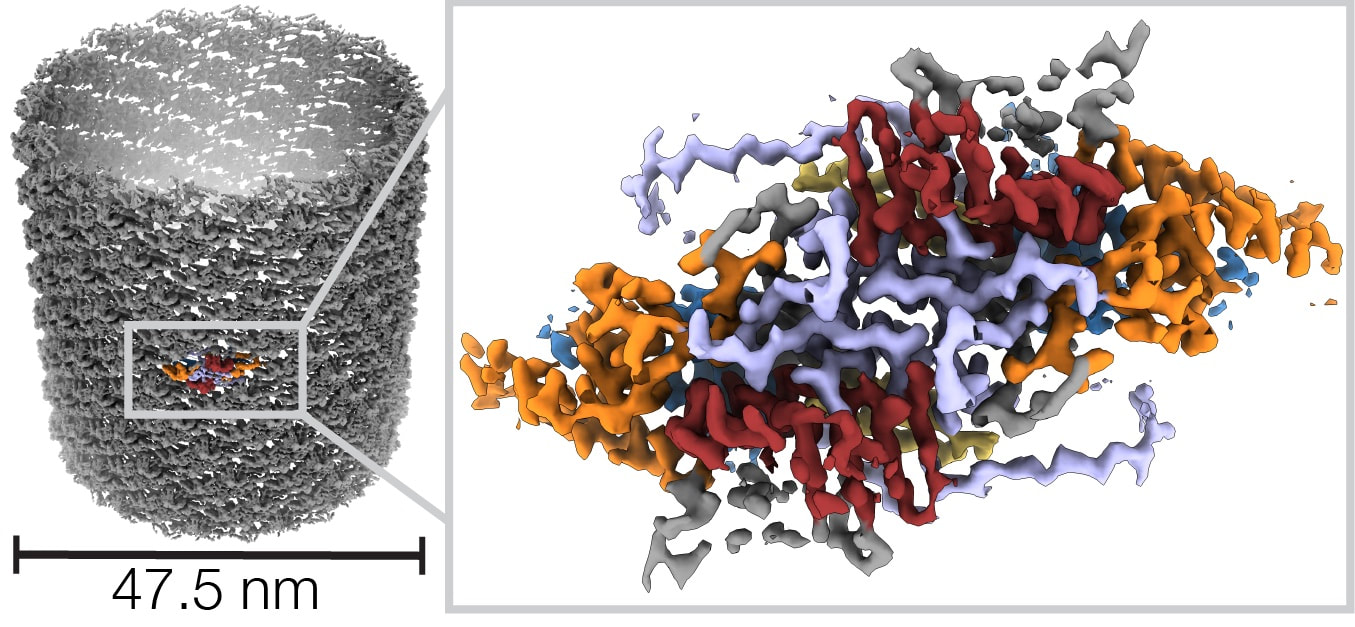
Helical reconstruction of VP39 reveals principles for baculovirus nucleocapsid assembly
Friederike M. C. Benning, Simon Jenni, Coby Y. Garcia, Tran H. Nguyen, Xuewu Zhang, Luke H. Chao Nature Communications 2024 Jan 4;15(1):250. doi: 10.1038/s41467-023-44596-y. PMID: 38177118. Baculoviruses are insect-infecting pathogens with wide applications as biological pesticides, in vitro protein production vehicles and gene therapy tools. Its cylindrical nucleocapsid, which encapsulates and protects the circular double-stranded viral DNA encoding proteins for viral replication and entry, is formed by the highly conserved major capsid protein VP39. The mechanism for VP39 assembly remains unknown. We determined a 3.2 Å electron cryomicroscopy helical reconstruction of an infectious nucleocapsid of Autographa californica multiple nucleopolyhedrovirus, revealing how dimers of VP39 assemble into a 14-stranded helical tube. We show that VP39 comprises a unique protein fold conserved across baculoviruses, which includes a Zinc finger domain and a stabilizing intra-dimer sling. Analysis of sample polymorphism revealed tube flattening could account for different helical geometries. This VP39 reconstruction reveals general principles for baculoviral nucleocapsid assembly. |
|
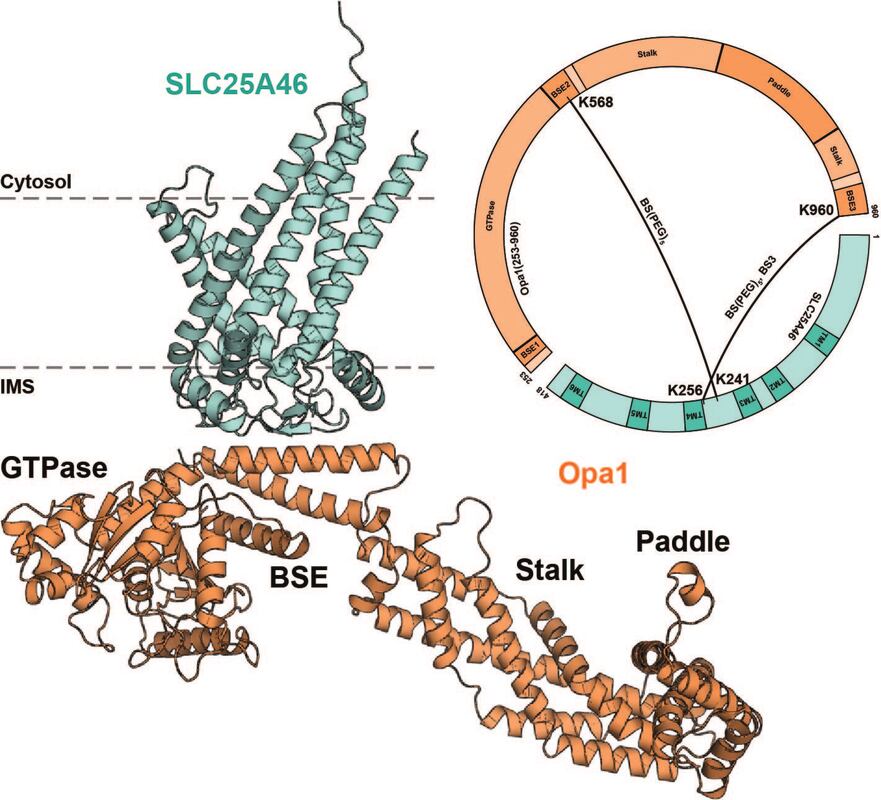
Identification of SLC25A46 interaction interfaces with mitochondrial membrane fusogens Mfn2 and Opa1
Sivakumar Boopathy, Camila Makhlouta Lugo, Bridget E Luce, Julie L McDonald, Pusparanee Hakim, Jackeline Ponce, Beatrix Ueberheide, Luke H Chao bioRxiv 2023.12.29.573615v1; doi: https://doi.org/10.1101/2023.12.29.573615 Mitochondrial fusion requires the sequential merger of four bilayers to two. The outer-membrane solute carrier protein SLC25A46 interacts with both the outer and inner-membrane dynamin family GTPases Mfn1/2 and Opa1. While SLC25A46 levels are known affect mitochondrial morphology, how SLC25A46 interacts with Mfn1/2 and Opa1 to regulate membrane fusion is not understood. In this study, we use crosslinking mass-spectrometry and AlphaFold 2 modeling to identify interfaces mediating a SLC25A46-Opa1-Mfn1/2 complex. We reveal that the bundle signaling element of Opa1 interacts with SLC25A46, and the helical repeat 1 region of Mfn2 interacts with the SLC25A46 N-terminus. We validate these newly identified interaction interfaces and show that they play a role in mitochondrial network maintenance. |
|
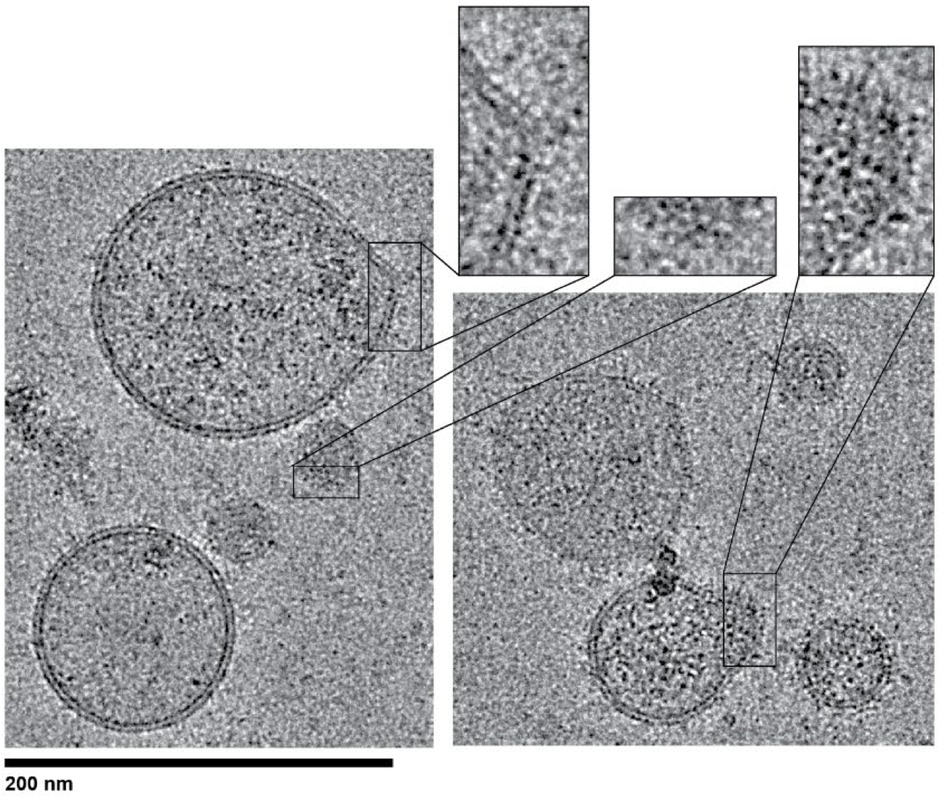
Cholesterol regulates plasma membrane bending by prominin-family proteins
Tristan A. Bell^, Bridget E. Luce, Pusparanee Hakim, Hiba Dardari, Virly Y. Ananda, Tran H. Nguyen, Arezu Monshizadeh, Luke H. Chao^ bioRxiv 2023.11.08.566258v2; doi: https://doi.org/10.1101/2023.11.08.566258 ^co-corresponding Prominin-1 (Prom1) is a pentaspan membrane protein that associates with curved regions of the plasma membrane. Prom1 localizes to cholesterol-rich domains and requires membrane cholesterol to support membrane remodeling. Membrane bending activity is particularly evident in photoreceptors, where Prom1 mutations cause loss of outer segment disk homeostasis leading to cone-rod retinal dystrophy (CCRD). However, the mechanistic link between prominin-dependent cholesterol binding, membrane remodeling, and retinal disease remains unclear. Here, we characterize the membrane bending function and specific cholesterol binding activity of Prom1 and its proposed homolog Tweety homology 1 (Ttyh1) in extracellular vesicles (EVs). Prom1 and Ttyh1 induce formation of EVs in cultured mammalian cells that are biophysically similar. Though both proteins bend membranes and form EVs at the plasma membrane, Ttyh1 lacks a stable interaction with cholesterol that is present in Prom1. Correspondingly, Ttyh1 forms EVs that are more deformed than those produced by Prom1. An evolutionarily conserved and retinal disease-associated Prom1 residue (Trp-795) is necessary for cholesterol binding, EV membrane deformation, and efficient trafficking to the plasma membrane. Removal of N-glycan moieties from Prom1 biases the enzyme toward a cholesterol-bound state. We propose that Prom1 and Ttyh1 are both members of a single prominin family of membrane bending proteins, that Ttyh1 is a constitutively active member of this family, and that Prom1 is regulated by cholesterol binding and N-glycosylation. These findings shed light on mechanisms of prominin family function in disease and help unify models of prominin function across diverse cell types. |

|
Cell wall synthesis and remodeling dynamics determine bacterial division site architecture and cell shape
Paula P. Navarro*, Andrea Vettiger*, Virly Y Ananda, Paula Montero Llopis, Christoph Allolio, Thomas G Bernhardt^, Luke H Chao^ Nature Microbiology, (2022). https://doi.org/10.1038/s41564-022-01210-z PDF *equal contribution ^co-corresponding The bacterial division apparatus catalyses the synthesis and remodelling of septal peptidoglycan (sPG) to build the cell wall layer that fortifies the daughter cell poles. Understanding of this essential process has been limited by the lack of native three-dimensional views of developing septa. Here, we apply state-of-the-art cryogenic electron tomography (cryo-ET) and fluorescence microscopy to visualize the division site architecture and sPG biogenesis dynamics of the Gram-negative bacterium Escherichia coli. We identify a wedge-like sPG structure that fortifies the ingrowing septum. Experiments with strains defective in sPG biogenesis revealed that the septal architecture and mode of division can be modified to more closely resemble that of other Gram-negative (Caulobacter crescentus) or Gram-positive (Staphylococcus aureus) bacteria, suggesting that a conserved mechanism underlies the formation of different septal morphologies. Finally, analysis of mutants impaired in amidase activation (ΔenvC ΔnlpD) showed that cell wall remodelling affects the placement and stability of the cytokinetic ring. Taken together, our results support a model in which competition between the cell elongation and division machineries determines the shape of cell constrictions and the poles they form. They also highlight how the activity of the division system can be modulated to help generate the diverse array of shapes observed in the bacterial domain. |
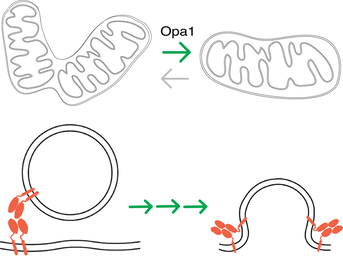
|
Two forms of Opa1 cooperate to complete fusion of the mitochondrial inner-membrane
Yifan Ge, Xiaojun Shi, Sivakumar Boopathy, Julie McDonald, Adam W Smith, Luke H Chao eLife 2020; 9:e50973 PMID: 31922487 PDF Mitochondrial membrane dynamics is a cellular rheostat that relates metabolic function and organelle morphology. Using an in vitro reconstitution system, we describe a mechanism for how mitochondrial inner-membrane fusion is regulated by the ratio of two forms of Opa1. We found that the long-form of Opa1 (l-Opa1) is sufficient for membrane docking, hemifusion and low levels of content release. However, stoichiometric levels of the processed, short form of Opa1 (s-Opa1) work together with l-Opa1 to mediate efficient and fast membrane pore opening. Additionally, we found that excess levels of s-Opa1 inhibit fusion activity, as seen under conditions of altered proteostasis. These observations describe a mechanism for gating membrane fusion. |